
Acidic Strength of Organic Acids
Share
Acidic Strength of Organic Acids
Fundamental of Organic Chemistry of Class 10
The factors responsible for influencing the acidity of an organic compound, H−X are
(a) The strength of the H − X bond
(b) The electronegativity of X
(c) Factors stabilizing X − as compared to HX/p
(d) The nature of the solvent.
The most important factors among all is (c). Now let us see using any of these factors, how we predict the order of acidic strength for organic acids.
(i) Alcohols: If we were to predict the order of acidic strength of ethanol, isopropanol and tertiary butanol, we can proceed as
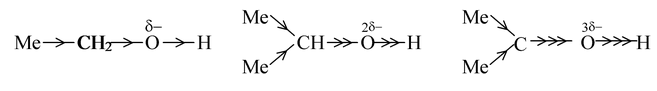
As alkyl groups (methyl group) have a +I effect, the electron density at the oxygen atom will increase. More the number of alkyl groups, more will be the intensity of negative charge on oxygen. The greater the negative charge on the oxygen atom, the closer is the electron pair in the O−H bond driven to the hydrogen atom and consequently separation of a proton becomes increasingly difficult. Thus, the acidic strength of given alcohols will be in the order
Ethanol > isopropanol > t−butanol
(ii) Phenols and substituted phenols: Phenols are stronger acid than alcohols. This is attributed to the fact that in phenoxide (anion obtained by the loss of proton from phenol), there is possibility of the delocalization of its negative charge through interaction with the π−orbitals of the aromatic nucleus while alkoxide cannot be stabilized by such effect. Thus phenoxide is a weaker base than alkoxide. Consequently, the phenol would be stronger acid than alcohol.
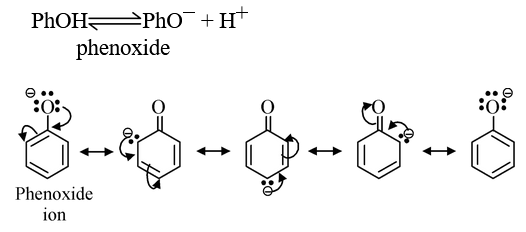
Delocalization also occurs in the undissociated phenol but as it involves charge separation, it is less effective than in the phenoxide. Thus, phenoxide shows some reluctance to take up proton and the equilibrium lies to the right, indicating greater stabilization of phenoxide and more acidity of phenols with respect to alcohols.
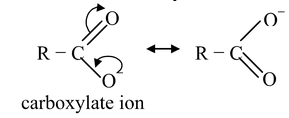
Phenols are less acidic than carboxylic acids due to the fact that delocalozation of the negative charge in the carboxylate anion involves structures of identical energy content (as the negative charge resides on more electronegative atom, oxygen) while in phenoxide, the negative charge also resides on the less electronegative atom (carbon) making these structures of high energy content with respect to those structures in which the negative charge is on oxygen. Thus, the relative stabilization of the carboxylate with respect to the undissociated carboxylic acid is more effective than the relative stabilization of phenoxide w.r.t. to undissociated phenol. Thus, carboxylate is a weaker base than phenoxide, making the corresponding carboxylic acid, a stronger acid than phenol.
R−CO
2
H
 RCO
-
2
+ H
+
RCO
-
2
+ H
+
carboxylate ion
The presence of electron−with drawing groups in phenol increases its acidity while the presence of electron releasing groups decreases the acidity of phenol.
Now, let us compare the acidic strength of o−, m− and p−nitrophenols. The corresponding phenoxide ions obtained from the three nitrophenols would be stabilized by delocalization as
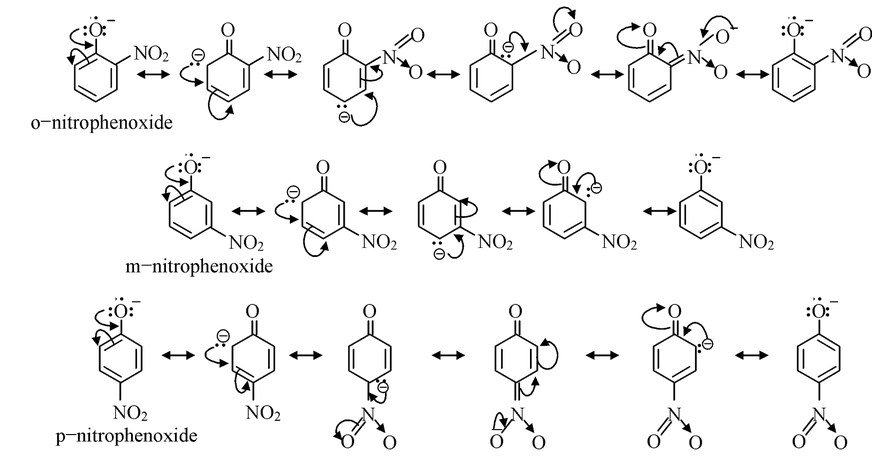
From the structures of phenoxide ions, it is evident that o−and p−nitrophenoxides are more extensively stabilized with respect to m−nitro phenoxide (as additional contributing structure is obtained by the dispersal of negative charge over oxygen atom of NO 2 group also in o− and p−nitrophenoxide but not in m−nitrophenoxide). Thus corresponding o− and p−nitrophenols are much stronger acids than m−nitrophenol. Out of o− and p−nitrophenols, p−nitrophenol is slightly stronger than o−isomer as o−isomer is bit stabilized by intramolecular hydrogen bonding, thus having a decreased tendency to release proton.
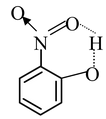
The order of acidic strength of the nitrophenols is
p−nitrophenol> o−nitrophenol> m−nitrophenol
Aliphatic carboxylic acids: The aliphatic carboxylic acids are much stronger acids than phenols and alcohols. This is attributed to the fact that the carboxylate ion (obtained by the loss of proton from carboxylic acid) is relatively more stabilized by delocalization than the phenoxide and alkoxide ions with respect to the their undissociated molecules.
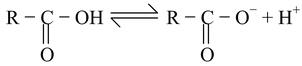
The presence of electron−with drawing substituents in simple aliphatic acids increases their acidity while the electron−releasing substituents have reverse effect. For instance, let us compare the acidic strength of fluoroacetic acid and acetic acid.
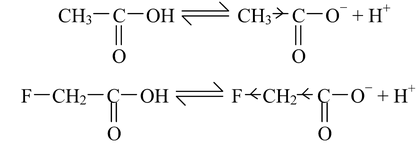
The fluoro acetate ion is stabilized more due to strong −I effect of fluorine, with respect to acetate ion. Hence, fluro acetate ion is less basic than acetate ion, thereby making fluoroacetic acid stronger than acetic acid.
If there is a doubly bonded carbon atom adjacent to the carboxyl group, the acid strength is increased. This will be evident if we compare the acid strength of propanoic acid and propenoic (acrylic) acid.
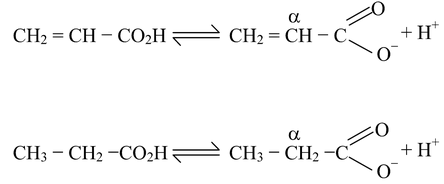
Basic Strength of Organic Bases
Strength of bases is related to the ease of accepting a proton which inturn depends on the availability of electron pair on the nitrogen atom (or some other basic atom). More is the availability of electron pair, more easily the proton will be accepted and more will be the basic strength.
(i) Aliphatic Bases
As increasing strength in nitrogenous bases is related to the readiness with which they take up protons and which inturn depends on the availability of the unshared electron pair on nitrogen, we expect an increase in basic strength on going from NH<>3→ RNH 2 → R 2 NH → R 3 N, due to the increasing inductive effect of successive alkyl groups making the nitrogen atom more electron rich.
But in actual practice the order of basic strength of amines is
NH 3 < Me 3 N < MeNH 2 < Me 2 NH
pKa's 9.25 9.80 10.64 10.77
NH 3 < EtNH 2 < Et 3 N < Et 2 NH
and 9.25 10.67 10.88 10.93
It can be seen that the introduction of an alkyl group into ammonia increases the basic strength markedly as expected. The introduction of a second alkyl group further increases the basic strength, but the net effect of introducing the second alkyl group is very much less marked than with the first. However, the introduction of a third alkyl group to yield a tertiary amine, actually decreases the basic strength. This is due to the fact that the basic strength of an amine in water is determined not only by electron−availability on the nitrogen atom, but also by the extent to which the cation, (formed by the uptake of a proton), can undergo solvation and so become stabilized. The more hydrogen atoms attached to nitrogen in the cation, the greater the possibilities of extensive solvation via hydrogen bonding between protonated amines and water.
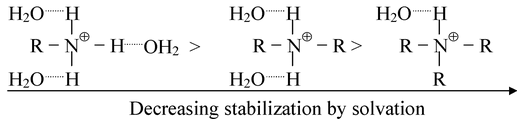
Thus on going along the series, NH 3 → RNH 2 → R 2 NH → R 3 N, the inductive effect will tend to increase the basicity but progressively less stabilization of the cation by hydration will tend to decrease the basicity. The net effect of introducing successive alkyl groups thus becomes progressively smaller, and an actual changeover takes place on going from a secondary to a tertiary amine. If this is assumed to be the right explanation, no such changeover should be observed if measurements of basicity are made in a solvent in which hydrogen−bonding cannot take place. This is indeed found to be true. In chlorobenzene, the order of basicity of the butylamines is BuNH 2 < Bu 2 NH < Bu 3 N while their basic strength in water follows the order Bu 3 N < BuNH 2 < Bu 2 NH.
Tetraalkylammonium salts, e.g.R 4 N ⊕ I - on treatment with moist silver oxide (AgOH) to yield basic solutions comparable in strength with the mineral alkalis. This is readily understandable as R 4 N ⊕− OH formed is completely ionized to give R 4 N + and free OH − .
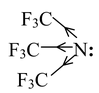
The effect of introducing electron−withdrawing groups, e.g. Cl, NO 2 , close to a basic centre decreases the basicity, due to their electron−withdrawing inductive effect. Thus the amine Tris (trifluoro methyl) amine is found to be virtually non−basic due to the presence of three powerfully electron−withdrawing CF 3 groups.
The amides are also found to be only very weakly basic in water [pK a for ethanamide(acetamide) is ≈ 0.5], because of the −I and −R effect of RCO group which makes the electron pair very slightly available on nitrogen atom.
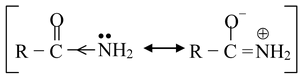
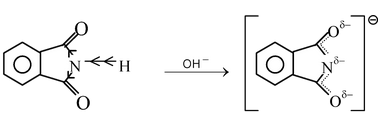
If two C = O groups are present the resultant imides often become sufficiently acidic to form alkali metal salts. For example, benzene−1,2−dicarboximide is not basic but is acidic in nature because of the presence of two electron-withdrawing CO groups.
Sometimes delocalisation also helps in increasing the basic strength of an amine, as seen in guanidine HN = C(NH 2 ) 2 . The guanidine is among the stronger bases known (organic nitrogenous) with a pK a of 1.36 (but less stronger than tetraalkyl ammonium hydroxides). Both the neutral molecule and the cation, H 2 N ⊕ = C(NH 2 ) 2 , resulting from its protonation are stabilized by delocalisation.










