

Peptide Bonds and Ramachandran Plots
Peptide Bonds and Ramachandran Plots: The results of the decoding process, which begins with the information in cellular DNA, are proteins. Proteins are the structural and motor components of the cell and operate as catalysts for almost all biochemical reactions that take place in living things, making them the cell's workhorses. A breathtakingly basic code that defines a tremendously varied collection of structures yields this amazing array of functions.
In fact, the DNA of every cell has genes that have code that encodes a certain protein structure. These proteins are constructed using various amino acid sequences, as well as various bonds that hold them together when they are folded into various three-dimensional structures. The protein's linear amino acid sequence directly affects the folded shape, or conformation.
What Constitutes Protein?
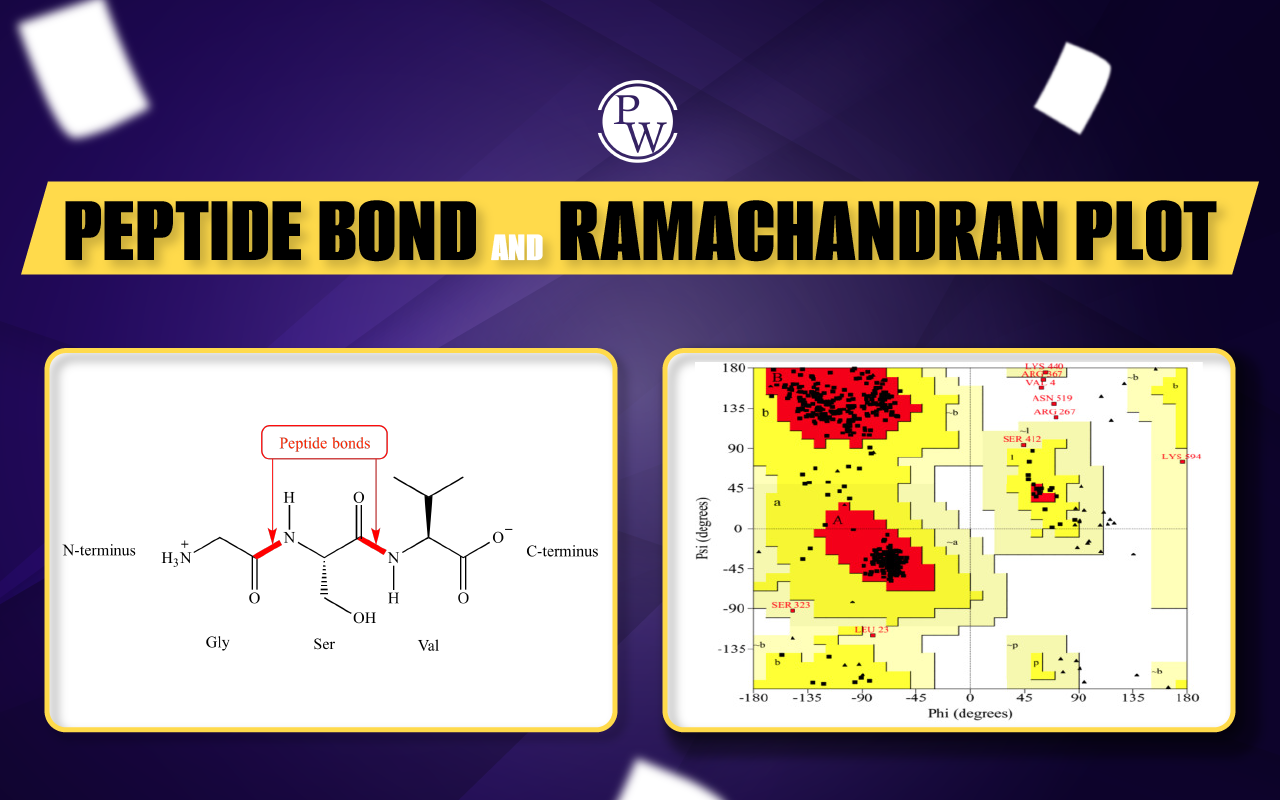
Amino acids, which are tiny chemical compounds with an alpha (central) carbon atom coupled to an amino group, a carboxyl group, a hydrogen atom, and a variable component known as a side chain, are the building blocks of proteins. A protein is made up of a lengthy chain of amino acids that are connected by peptide bonds. Peptide bonds are formed by a biochemical reaction that extracts a water molecule as it joins the amino group of one amino acid to the carboxyl group of a neighboring amino acid. The primary structure of a protein is thought to be its linear amino acid sequence.
Only twenty different amino acids, each with a distinct side chain, make up proteins. Different chemistry can be found in the side chains of amino acids. The majority of amino acids have side chains that are nonpolar. While some amino acids have polar but uncharged side chains, others have side chains with positive or negative charges. Because amino acid side chains can form bonds with one another to retain a length of protein in a certain shape or conformation, their chemistry is essential to protein structure. Ionic connections can develop between charged amino acid side chains, and hydrogen bonds can form between polar amino acids. Weak van der Waals interactions mediate interactions between hydrophobic side chains. These side chains create a large number of noncovalent bonds. Actually, due to their unique side chains, cysteines are the only amino acids that can create covalent connections. Because of side chain interactions, a protein's amino acid sequence and position determine where its bends and folds take place.
The primary structure of a protein, its amino acid sequence , drives the folding and intramolecular bonding of the linear amino acid chain, which ultimately determines the protein's unique three-dimensional shape. Hydrogen bonding between amino groups and carboxyl groups in neighboring regions of the protein chain sometimes causes certain patterns of folding to occur. Known as alpha helices and beta sheets, these stable folding patterns make up the secondary structure of a protein. Most proteins contain multiple helices and sheets, in addition to other less common patterns. The ensemble of formations and folds in a single linear chain of amino acids, sometimes called a polypeptide, constitutes the tertiary structure of a protein. Finally, the quaternary structure of a protein refers to those macromolecules with multiple polypeptide chains or subunits.
Partial Double Bond Character
In an organic molecule, resonance results in partial double bonds. The partial double bond feature results from the pi electrons moving from one p-orbital to another. A peptide has a double bond partial character because it shares an electron in the peptide bond between the oxygen and the nitrogen. This partial double bond of the peptide bond results in the rigid and planar properties of the peptide bond. The peptide bond in proteins is most often in the trans configuration.
Ramachandran Plot
A Ramachandran plot was first created by Viswanathan Sasisekharan (born in 1993), C. Ramakrishnan, and Gopalasamudram Narayana Ramachandran. It is also referred to as a Ramachandran diagram or a Rama plot in the field of biochemistry. The plot of angles known as psi and phi of the residues—commonly referred to as amino acids—present in a peptide is known as a Ramachandran plot. Ramachandran plot is a method for imagining enthusiastically (or) energetically permitted areas for backbone or spine dihedral points ψ against φ of amino acid buildups in the protein structure. As the partial-double-bond keeps the peptide bond planar, the ω angle at that particular peptide bond is always 180 degrees (180°). Numerous combinations of angles as well as residue conformations are not feasible due to steric hindrance. This Ramachandran plot can be used to establish the acceptable angles for gaining insight into the structure of proteins.
The main chain N- and C-bonds are largely free to spin in polypeptides. The torsion angles phi and psi, respectively, stand in for these rotations. In order to find stable conformations, G. N. Ramachandran used computer models of tiny polypeptides to repeatedly change phi and psi. The structure was analyzed for atom-atom interactions in each configuration. Atoms were viewed as solid spheres with van der Waals radii-based dimensions. As a result, phi and psi angles that cause spheres to collide correspond to conformations of the polypeptide backbone that are sterically forbidden.
Regions in Ramachandran Plot
Ramachandran plots can be used to determine the secondary structure of proteins. The plot of Ramachandran is divided into four quadrants.
- Quadrant-I: Quadrant-I is the area of confirmations, where all the confirmations are allowed. In this region, we can find left-handed alpha.
- Quadrant-II: Quadrant-II is the biggest region in the whole graph. Particularly, this region has better conditions for the confirmation of atoms.
- Quadrant-III: Quadrant-III is the biggest region after Quadrant-II. In this region, we can find right-handed alpha.
- Quadrant-IV: Quadrant-IV has practically no framed locale. This conformation (ψ around—180 to 0 degrees, φ around 0-180 degrees) is disfavored due to steric conflict.
The white areas in the diagram above represent conformations in which polypeptide atoms are closer together than the total of their van der Waals radii. With the exception of glycine, which is distinct in that it doesn't have a side chain, these locations are sterically forbidden for all amino acids. The alpha-helical and beta-sheet conformations are the permissible zones, and the red regions correspond to conformations in which there are no steric collisions. If a little shorter van der Waals radii are used in the computation or if the atoms are permitted to get a bit closer together, the yellow areas represent the permitted zones. The left-handed alpha-helix's extra area is revealed as a result.
Although extended left-handed helix sections cannot be formed by L-amino acids, occasionally a single residue will take on this shape. These residues, which are often glycine but may also be asparagine or aspartate when the side chain interacts with the main chain to establish a hydrogen bond, stabilize this otherwise unfavorable shape. Close to the upper right of the alpha-helical zone, the 3 10 helix is on the border of the permitted range, indicating lesser stability.
L-amino acids cannot form extended regions of a left-handed helix, but occasionally individual residues adopt this conformation. These residues are usually glycine but can also be asparagine or aspartate, where the side chain forms a hydrogen bond with the main chain and therefore stabilizes this otherwise unfavorable conformation. The 3 10 helix occurs close to the upper right of the alpha-helical region and is on the edge of the allowed region, indicating lower stability. Disallowed regions generally involve steric hindrance between the side chain C-beta methylene group and main chain atoms. Glycine has no side chain and therefore can adopt phi and psi angles in all four quadrants of the Ramachandran plot. Hence, it frequently occurs in turn regions of proteins where any other residue would be sterically hindered.
Uses
A Ramachandran plot can be utilized in two various ways. One is to show in principle which values, or conformities, of the psi(ψ) and phi(φ) torsional angles, are feasible for the buildup or residue of an amino-corrosive in a protein structure. This is the first advantage of this plot. The second one is to show the observational dissemination of data points seen in a solitary design in utilization for structure approval, or, more than likely, in a data set of many designs. Either body of evidence is normally displayed against frames for the hypothetically preferred locales.












