
Aliphatic Nucleophilic Substitution
Alkyl and Aryl Halides of Class 12
In nucleophilic substitutions, the attacking reagent (nucleophile) brings an electron pair to the substrate, which uses this pair to form a new bond and the leaving group comes away with an electron pair. When nucleophile is a solvent, the reaction is called solvolysis.
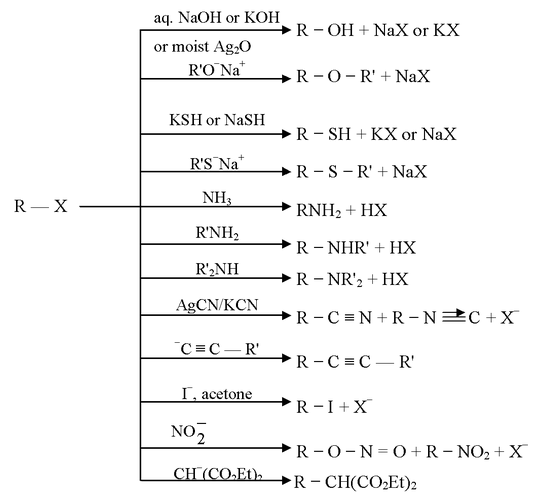
R − X + Y  R − Y + X
R − Y + X
Y is a nucleophile which may be neutral or negatively charged while RX may be neutral or positively charged.
R − I + HO− R − OH + I−
R − I + NMe3 R−  + I−
+ I−
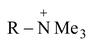 + OH− R − OH + NMe3
+ OH− R − OH + NMe3
+ H2S R − s+H2 +NMe3
Before going on for discussion of the mechanism of nucleophilic substitution reactions in alkyl halides, we must have an idea about the few basic terms. These are mentioned below.
Basicity and Nucleophilicity
Basicityis the tendency of a base of abstract H+ and nucleophilicity is its tendency to attack as a nucleophileon carbon. All the bases are nucleophiles and all the nucleophiles are bases.
SN1 REACTION
SN1 stands for substitution nucleophilic unimolecular. The ideal version of SN1 consists of two steps. The first step is the slow ionization of the substrate and is rate determining step. The second step is the rapid reaction between the carbocation and nucleophile.
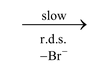
R − X 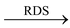 R+ + X−Step 1 (slow)
R+ + X−Step 1 (slow)
R+ + Y− R − YStep 2 (fast)
The ionization is always assisted by solvent, since the energy of activation required for breaking of the bond is largely recovered from solvation of ions produced. For example, ionization of t−BuCl in gas phase requires about 150 kcal/mol but in water it requires only 20 kcal/mol. In pure SN1 reactions, solvent molecules assist the departure of leaving group from the front side.
The carbocation generated by first step has an Sp2 hybridized carbon i.e. the structure is flat (trigonal planar). Thus nucleophile will attack the carbocation from the front side as well as from the rear side with equal ease, leading to the formation of two isomers, if the chiral carbon is present in the substrate.
Stereochemical Aspects of SN1 Mechanism
Let us understand the mechanistic details of SN1 reaction in the following manner:
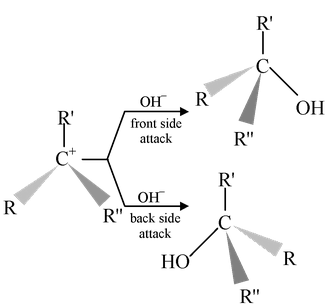
A planar carbocation is formed during the slow rds of the reaction, thus if may be expected that the nucleophilic attack by OH− (or solvent H2O) would occur from either side of the carbocation leading to the formation of 50/50 mixture of substituted products having same and opposite configuration as that of starting material, i.e. racemisation will occur leading to an optically inactive (±) product.
In practice, the complete racemisation is never observed. It is always accompanied by some degree of inversion. The relative proportion of the two depend on (a) the stability of carbocation (b) the ability of solvent as nucleophile (c) the ionizing ability of the solvent and (d) the bulkiness of the groups on the substrate.
Till the formation of free carbocation, the nucleophile attacks from the rear side of the compound leading to inverted product while once the carbocation is formed the probability of attack from both sides are equally feasible. As a result, overall inverted product exceeds the retented product.
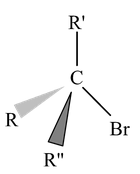
SN2 Reaction Mechanism
SN2 stands for substitution nucleophilic bimolecular. In this mechanism, there is a backside attack of the nucleophile. The nucleophile approaches the substrate from a position 180° away from the leaving group. The reaction is a one−step process with no intermediate. The C − Y bond is formed as C − X bond is broken.
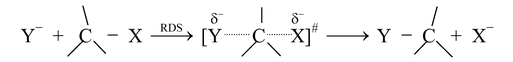
Transition State
The energy necessary to break the C−X bond is supplied by the simultaneous formation of
C− Y bond. The position of atoms at the top of the curve of activation energy can be represented as Transition state (TS). The group X must leave as Y comes in, because at no time can the carbon atom have more than 8 electrons in its outermost shell.
In the transition state, the carbon is sp2 hybridized with a p atomic orbital available for overlapping of its lobes with an orbital of the incoming :Nu− while the other lobe overlaps with an orbital of the leaving group X−. These overlaps account for the partial bonds drawn in figure. The reaction is initiated by :Nu− beginning to overlap with the small lobe (tail) of the sp3 hybrid orbital bonding with X. In order to provide more bonding volume to give a stronger bond, the tail becomes the larger lobe (head) and the head becomes the tail, inverting the configuration of carbon. The configuration of the original compound is the opposite to that of the compound obtained. As :Nu− starts to bond to carbon, it loses some of its full charge and in the transition state has a δ− charge, as does X as it begins to leave as an anion.
In SN2 mechanism, the front side attack has never been observed. In a hypothetical front side attack, both the nucleophile and nucleofuge (leaving group) would have to overlap with the same lobe of p−orbital whereas the back side attack involves the maximum amount of overlap throughout the course of reaction. During the transition state, the three non−reacting groups and the central carbon atom are approximately coplanar.
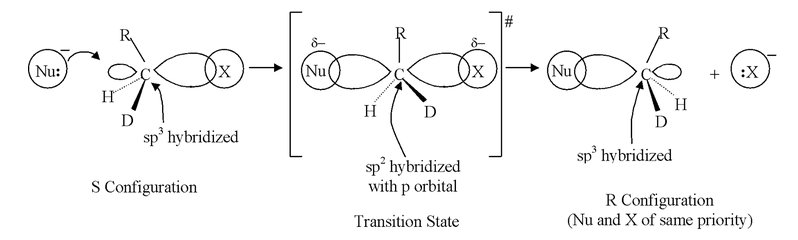
In the above representation it is clear that the carbon atom is linked to five groups so it is highly overcrowded. Due to high overcrowding it is easier to make a decision that more bigger the groups attached to carbon, more unstable will be the transition state. Hence steric factors will have important role in SN2 substitution. Due to this reason the reactivity towards SN2 is as follows.
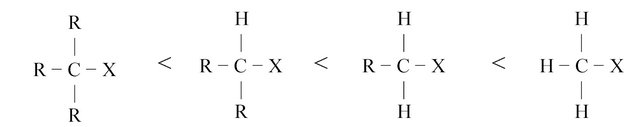
SN2 reactions are stereospecific because stereoisomeric reactants give stereochemically different products. They are also stereoselective because they form exclusively or predominantly only one of a possible pair of enantiomers or one of the possible diastereoisomers.
1. Comparative Details of SN1 and SN2 reactions
|
SN1 |
SN2 |
|
|
(a) Number of steps |
2 steps
(i) R : L
(II) R+ + : NuH |
1 step R : L + : Nu−→ R : Nu +: L− OR R : L + : NuH → R : N+uH+: L− |
|
(b) Reaction rate & order |
Rate = k1[RL]; first order |
Rate =k1[RL][:Nu−]; second order |
|
(c) Molecularity |
Unimolecular |
Bimolecular |
|
(d) TS of slow step |
HNu: ……. Cδ+……. δ− L ……. HNu: |
δ−Nu ……. C ……. :Lδ− (with : Nu−) δ+HNu…….C…….:Lδ−(with :Hnu) |
|
(e) Stereochemistry |
Inversion and retention (Partial racemization) |
Inversion (backside attack) |
|
(f) Reacting nucleophile |
Nucleophilic solvent; stable R+ may react with added nucleophile |
Added nucleophile |
|
(g) Structure of R |
3° > 2° > 1° > Me |
Me > 1° > 2° > 3° |
|
(h) Nature of Leaving group |
Weakest base is best leaving group, i.e. |
Weakest base is best leaving group, i.e. I− > Br− > Cl− > F− |
|
(i) Solvent effect |
Rate α H−bonding ability and dielectric constant |
Depends on charge type. Polar aprotic solvents leaves "freest" most reactive Nu. |
|
(j) Determining factor |
Stability of R+ |
Steric hindrance |
|
(k) Rearrangement |
Observed |
Not observed, except for allylic |
|
(l) Catalysis |
Lewis and Bronsted acids:Ag+, AlCl3 and strong HA |
No specific catalyst |
 R+ + : L−
R+ + : L− R:Nu + H+
R:Nu + H+-
Ambident Nucleophiles
Some nucleophiles have lone pair of electrons on more than one atom and can attack through more than one site. Such nucleophiles are called ambident nucleophiles. In such cases different products due to attack by different sites are possible. Attack by a specific site can be promoted under special conditions. Two well known examples are discussed in detail.
Attack by CN− nucleophile (:−C ≡ N :)
R − X  R − CN + R − NC + X−
R − CN + R − NC + X−
nitrilesisonitriles
In CN−, carbon (negatively charged) will be a soft base as compared to nitrogen. So if the reaction proceeds via SN1 mechanism, which produces a free carbocation (a hard acid), then attack through nitrogen (hard base) will take place. But if the reaction proceeds via SN2 mechanism (small positively charged carbon is soft acid) then attack through carbon (soft base) will take place. So if we want to increase relative yield of nitriles, we can use NaCNor KCN etc in a less polar solvent which facilitates SN2 substitution. Similarly if we want to increase the yield of isonitriles, we use AgCN. Ag+ has very high affinity for X− so it favours the formation of R+ and the reaction proceeds via SN1 mechanism. This will result in attack by hard base giving R−NC. Further if we compare primary, secondary and tertiary alkyl halides, formation of R − NC should be favoured due to more favourable SN1 substitution in tertiary alkyl halide. But the exception is that tertiary alkyl halides undergo elimination and the yield decreases. This is because CN− is a strong base which can cause elimination reaction easily.
Attack by  nucleophile (−O − N = O)
nucleophile (−O − N = O)
R − X 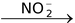 R − O − N = O+R − NO2 + X−
R − O − N = O+R − NO2 + X−
alkane nitritenitro alkane
In , oxygen (negatively charged) will be a hard base as compared to nitrogen. So if the reaction proceeds via SN1 mechanism, then attack through oxygen (hard base) will take place to produce alkane nitrite. But if the reaction takes place via SN2 mechanism then attack through nitrogen (soft base) takes place to give nitro alkane.
If we want to increase the yield of nitro alkane the reaction should proceed via SN2 mechanism. i.e. we can use NaNO2, KNO2 etc. Moreover the yield will be best if we use primary alkyl halide and less polar solvent. Formation of nitrite will dominate if we use tertiary alkyl halide, more polar solvent and AgNO2 because Ag+ has strong affinity for X− and can form a carbocation to force the reaction to proceed via SN1 mechanism. Primary alkyl halide with AgNO2 chiefly gives nitroalkane but if secondary and tertiary alkyl halides are used then AgNO2 will yield nitrite as the major product.
